無聲惡化的肺病 認識特發性肺纖維化(IPF)
第一手醫學
疾病簡介
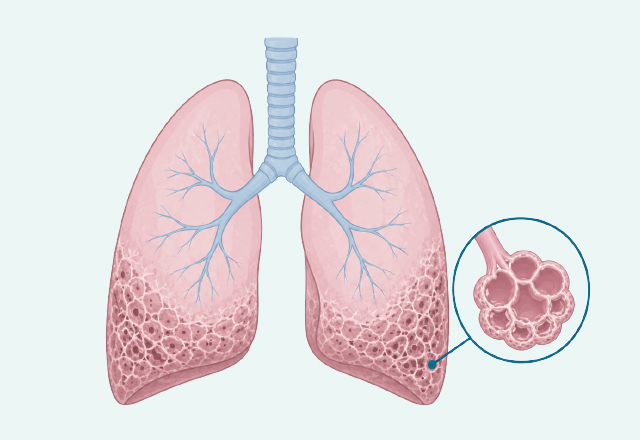
特發性肺纖維化(Idiopathic Pulmonary Fibrosis, IPF)是慢性且進展性的纖維化肺部疾病,其特徵在於肺部間質產生永久性的疤痕(纖維化)。這種疾病對患者的生活品質有顯著的負面影響,並會導致呼吸困難與咳嗽加劇。若未經治療,IPF 的進展十分迅速,患者的中位存活期僅約 3 年。
流行病學與疾病負擔
從流行病學角度來看,全球受影響人數約為 300 萬人。其發病率與盛行率隨著年齡增長而顯著增加,主要影響老年族群。根據研究,不同地區的發病率有所差異,例如亞太地區每萬人的發病估計值為 0.35 至 1.30,而北美地區則為0.75 至 0.93。
疾病成因與發病機制
IPF的確切發病機制尚未完全明確,但目前廣為接受的觀念是:肺泡上皮細胞(特別是第二型肺泡上皮細胞,AT2s)受到重複性的微小損傷,導致修復過程失調。
1.病理過程:這種受損引發了纖維母細胞(fibroblast)的遷移與積聚,並轉化為具活性的肌纖維母細胞(myofibroblast)。這些細胞過度分泌細胞外基質(ECM),最終導致肺部結構的重塑與廣泛瘢痕化。
2.遺傳因素:遺傳變異被認為可解釋約三分之一的疾病風險。其中,MUC5B 基因啟動子區域的變異(rs35705950)與發病風險最為相關,攜帶此變異者的患病機率可增加五倍。此外,端粒相關基因(如 TERC, TERT)的變異也與家族性及散發性 IPF 相關。
3.環境與危險因子:吸菸、環境暴露(如空氣污染、職業暴露)、慢性病毒感染(如人類疱疹病毒)以及胃食道逆流(GERD)均被視為潛在的危險因子。
臨床表現與診斷方法
IPF的常見症狀包括慢性乾咳與活動性呼吸困難。由於症狀與其他肺部疾病相似,診斷延遲相當常見,從症狀出現到最終診斷的中位時間約為 0.6 至 2.3年。
診斷流程主要結合臨床特徵、放射影像與病理組織:
•多學科團隊討論(MDT):這是目前診斷的「金標準」,由肺科醫師、放射科醫師及病理科醫師共同商議。
•高解析度電腦斷層(HRCT):這是核心診斷工具,典型的特徵稱為尋常性間質性肺炎(UIP)影像型態。
•排除其他成因:必須排除結締組織疾病(CTD)、藥物毒性或環境暴露引發的繼發性肺纖維化。必要時會進行肺部切片檢驗。
疾病管理與治療
目前 IPF 尚無法痊癒,治療目標在於延緩肺功能下降、減少急性惡化並提升生活品質。
1.藥物治療
目前已核准兩種抗纖維化藥物,能使肺功能(FVC)的下降速率減緩約50%:
•Nintedanib:一種酪氨酸激酶抑制劑,阻斷與纖維化相關的生長因子路徑。常見副作用為腹瀉,需監測肝功能。
•Pirfenidone:具抗炎與抗纖維化作用,能抑制膠原蛋白合成。常見副作用包括噁心、嘔吐及光敏性皮疹。
2. 非藥物管理
•氧氣治療:減輕呼吸困難並改善運動耐受力。
•肺復健:結構化的運動計畫可提升功能性生活品質。
•肺移植:對於符合條件的晚期患者,肺移植是延長生命的有效手段,目前IPF 是肺移植最常見的適應症。
•支持性與安寧照護:預先醫療計畫(ACP)對於幫助患者面對疾病末期至關重要。
未來展望與研發中藥物
科學界正積極研發針對不同致病路徑的新藥,包括:
•Pamrevlumab:針對結締組織生長因子的單株抗體,目前正進行第三期試驗。
•BI1015550:一種 PDE4B 抑制劑,初步研究顯示能有效阻止肺功能下降。
•其他路徑:如 Pentraxin-2、Galectin-3 抑制劑等亦在臨床試驗中展現潛力。
總結來說,IPF 是一種病程嚴重的罕見疾病,雖然目前的治療僅能延緩進展而非根治,但隨著單細胞測序技術與深度學習影像分析的進步,未來有望開發出更精準的個人化治療方案。對於病友而言,及早診斷、規律服藥並配合肺復健,是目前管理疾病的最佳策略。
參考資料:
Koudstaal T, Wijsenbeek MS. Idiopathic pulmonary fibrosis. Presse Med. 2023 Sep;52(3):104166.